This page was imported and needs to be de-wikified. Books should use wikilinks rather sparsely, and only to reference technical or esoteric terms that are critical to understanding the content. Most if not all wikilinks should simply be removed. Please remove {{dewikify}} after the page is dewikified.
In animals, when there is an oversupply of dietary carbohydrate, the excess carbohydrate is converted to triacylglycerol. This involves the synthesis of fatty acids from acetyl-CoA and the esterification of fatty acids in the production of triacylglycerol, a process called lipogenesis. Fatty acids are made by fatty acid synthases that polymerize and then reduce acetyl-CoA units. The acyl chains in the fatty acids are extended by a cycle of reactions that add the acetyl group, reduce it to an alcohol, dehydrate it to an alkene group and then reduce it again to an alkane group. The enzymes of fatty acid biosynthesis are divided into two groups, in animals and fungi all these fatty acid synthase reactions are carried out by a single multifunctional protein, while in plant plastids and bacteria separate enzymes perform each step in the pathway. The fatty acids may be subsequently converted to triacylglycerols that are packaged in lipoproteins and secreted from the liver.
The synthesis of unsaturated fatty acids involves a desaturation reaction, whereby a double bond is introduced into the fatty acyl chain. For example, in humans, the desaturation of stearic acid by stearoyl-CoA desaturase-1 produces oleic acid. The doubly unsaturated fatty acid linoleic acid as well as the triply unsaturated α-linolenic acid cannot be synthesized in mammalian tissues, and are therefore essential fatty acids and must be obtained from the diet.
Triacylglycerol synthesis takes place in the endoplasmic reticulum by metabolic pathways in which acyl groups in fatty acyl-CoAs are transferred to the hydroxyl groups of glycerol-3-phosphate and diacylglycerol.
Terpenes and isoprenoids, including the carotenoids, are made by the assembly and modification of isoprene units donated from the reactive precursors isopentenyl pyrophosphate and dimethylallyl pyrophosphate. These precursors can be made in different ways. In animals and archaea, the mevalonate pathway produces these compounds from acetyl-CoA, while in plants and bacteria the non-mevalonate pathway uses pyruvate and glyceraldehyde 3-phosphate as substrates. One important reaction that uses these activated isoprene donors is steroid biosynthesis. Here, the isoprene units are joined together to make squalene and then folded up and formed into a set of rings to make lanosterol. Lanosterol can then be converted into other steroids such as cholesterol and ergosterol[1].
This page may need to verify facts by citing reliable publications. You can help by adding references to reliable publications, or by correcting statements cited as fact.
Steroid biosynthesis is an anabolic metabolic pathway that produces steroids from simple precursors. This pathway is carried out in different ways in animals than in many other organisms, making the pathway a common target for antibiotics and other anti-infective drugs. In addition, steroid metabolism in humans is the target of cholesterol-lowering drugs such as statins.
It starts in the mevalonate pathway in humans, with Acetyl-CoA as building blocks, which form DMAPP and IPP. In following steps, DMAPP and IPP form lanosterol, the first steroid. Further modification belongs to the succeeding steroidogenesis.
Steroidogenesis is the biological process by which steroids are generated from cholesterol and transformed into other steroids. The pathways of steroidogenesis differ between different species, but the pathways of human steroidogenesis are shown in the figure.
Products of steroidogenesis include:
androgens
testosterone
estrogens and progesterone
corticoids
cortisol
aldosterone
Biosynthesis of testosterone
Like other steroid hormones, testosterone is derived from cholesterol (see figure to the right). The first step in the biosynthesis involves the oxidative cleavage of the sidechain of cholesterol by CYP11A, a mitochondrial cytochrome P450 oxidase with the loss of six carbon atoms to give pregnenolone. In the next step, two additional carbon atoms are removed by the CYP17A enzyme in the endoplasmic reticulum to yield a variety of C19 steroids. In addition, the 3-hydroxyl group is oxidized by 3-β-HSD to produce androstenedione. In the final and rate limiting step, the C-17 keto group androstenedione is reduced by 17-β hydroxysteroid dehydrogenase to yield testosterone.
The largest amounts of testosterone (>95%) are produced by the testes in men. It is also synthesized in far smaller quantities in women by the thecal cells of the ovaries, by the placenta, as well as by the zona reticularis of the adrenal cortex in both sexes. In the testes, testosterone is produced by the Leydig cells. The male generative glands also contain Sertoli cells which require testosterone for spermatogenesis. Like most hormones, testosterone is supplied to target tissues in the blood where much of it is transported bound to a specific plasma protein, sex hormone binding globulin (SHBG)[2].
Anabolic steroids and its misuse Anabolic steroids (AAS) were first isolated, identified and synthesized in the 1930s, and are now used therapeutically in medicine to stimulate bone growth and appetite, induce male puberty, and treat chronic wasting conditions, such as cancer and AIDS. Anabolic steroids also produce increases in muscle mass and physical strength, and are consequently used in sport and bodybuilding to enhance strength or physique. The known side effects include harmful changes in cholesterol levels (increased Low density lipoprotein and decreased High density lipoprotein), acne, high blood pressure, liver damage. Some of these effects can be mitigated by taking supplemental drugs.
AAS user in sports began in October 1954, John Ziegler, a doctor who treated American athletes, went to Vienna with the American weightlifting team. There he met a Russian physicist who, over "a few drinks", repeatedly asked "What are you giving your boys?" When Ziegler returned the question, the Russian said that his own athletes were being given testosterone.
Returning to America, Ziegler tried weak doses of testosterone on himself, on the American trainer Bob Hoffman and on two lifters, Jim Park and Yaz Kuzahara. All gained more weight and strength than any training programme would produce but there were side-effects. Ziegler sought a drug without after-effects and hit on an anabolic steroid, methandrostenolone, (Dianabol, DBOL), made in the US in 1958 by Ciba.
The results were impressive—so impressive that lifters began taking ever more. Steroids spread to other sports where bulk mattered. Paul Lowe, a former running back with the San Diego Chargers American football team, told a California legislative committee on drug abuse in 1970: "We had to take them [steroids] at lunchtime. He [an official] would put them on a little saucer and prescribed them for us to take them and if not he would suggest there might be a fine."
Olympic statistics show the weight of shot putters grew 14 per cent between 1956 and 1972, whereas steeplechasers grew 7.6 per cent. The gold medallist pentathlete Mary Peters said: "A medical research team in the United States attempted to set up extensive research into the effects of steroids on weightlifters and throwers, only to discover that there were so few who weren't taking them that they couldn't establish any worthwhile comparisons."
In 1984, Jay Silvester, a former four-time Olympian and 1972 silver medalist in the discus who was then with the physical education department of Brigham Young University in the U.S., questioned competitors at that year's Olympics. The range of steroid use he found ranged from 10 mg a day to 100 mg[3].
A famous case of illicit AAS use in a competition was Canadian Ben Johnson's victory in the 100 m at the 1988 Summer Olympics. He subsequently failed the drug test when stanozolol was found in his urine. He later admitted to using the steroid as well as Dianabol, Testosterone Cypionate, Furazabol, and human growth hormone amongst other things. Johnson was therefore stripped of his gold medal as well as recognition of what had been a world-record performance. Carl Lewis was then promoted one place to take the Olympic gold title. Lewis had also run under the current world record time and was therefore recognized as the new record holder, however, in 2003, Dr. Wade Exum, the United States Olympic Committee (USOC) director of drug control administration from 1991 to 2000, gave copies of documents to Sports Illustrated which revealed that some 100 American athletes who failed drug tests and should have been prevented from competing in the Olympics were nevertheless cleared to compete. Among those athletes was Carl Lewis. Lewis then broke his silence on allegations that he was the beneficiary of a drugs cover-up, admitting he had tested positive for banned substances but claiming he was just one of "hundreds" of American athletes who were allowed to escape bans, concealed by the USOC. Lewis has now acknowledged that he failed three tests during the 1988 US Olympic trials, which under international rules at the time should have prevented him from competing in the Seoul games two months later.
Former athletes and officials came out against the USOC cover-up. "For so many years I lived it. I knew this was going on, but there's absolutely nothing you can do as an athlete. You have to believe governing bodies are doing what they are supposed to do. And it is obvious they did not", said former American sprinter and 1984 Olympic champion, Evelyn Ashford.
Biosynthesis of cortisol
Cortisol is synthesized from cholesterol. Synthesis takes place in the zona fasciculata of the adrenal cortex. (The name cortisol is derived from cortex.) While the adrenal cortex also produces aldosterone (in the zona glomerulosa) and some sex hormones (in the zona reticularis), cortisol is its main secretion. The medulla of the adrenal gland lies under the cortex, mainly secreting the catecholamines adrenaline (epinephrine) and noradrenaline (norepinephrine) under sympathetic stimulation.
The synthesis of cortisol in the adrenal gland is stimulated by the anterior lobe of the pituitary gland with adrenocorticotropic hormone (ACTH); ACTH production is in turn stimulated by corticotropin-releasing hormone (CRH), which is released by the hypothalamus. ACTH increases the concentration of cholesterol in the inner mitochondrial membrane, via regulation of the STAR (steroidogenic acute regulatory) protein. It also stimulates the main rate-limiting step in cortisol synthesis, in which cholesterol is converted to pregnenolone and catalyzed by Cytochrome P450SCC (side chain cleavage enzyme).
All animal cells manufacture cholesterol with relative production rates varying by cell type and organ function. About 20–25% of total daily cholesterol production occurs in the liver; other sites of higher synthesis rates include the intestines, adrenal glands, and reproductive organs. Synthesis within the body starts with one molecule of acetyl CoA and one molecule of acetoacetyl-CoA, which are dehydrated to form 3-hydroxy-3-methylglutaryl CoA (HMG-CoA). This molecule is then reduced to mevalonate by the enzyme HMG-CoA reductase. This step is the regulated, rate-limiting and irreversible step in cholesterol synthesis and is the site of action for the statin drugs (HMG-CoA reductase competitive inhibitors).
Mevalonate is then converted to 3-isopentenyl pyrophosphate in three reactions that require ATP. This molecule is decarboxylated to isopentenyl pyrophosphate, which is a key metabolite for various biological reactions. Three molecules of isopentenyl pyrophosphate condense to form farnesyl pyrophosphate through the action of geranyl transferase. Two molecules of farnesyl pyrophosphate then condense to form squalene by the action of squalene synthase in the endoplasmic reticulum. Oxidosqualene cyclase then cyclizes squalene to form lanosterol. Finally, lanosterol is then converted to cholesterol.
Konrad Bloch and Feodor Lynen shared the Nobel Prize in Physiology or Medicine in 1964 for their discoveries concerning the mechanism and regulation of cholesterol and fatty acid metabolism[4].
Biosynthesis of cholesterol is directly regulated by the cholesterol levels present, though the homeostatic mechanisms involved are only partly understood. A higher intake from food leads to a net decrease in endogenous production, whereas lower intake from food has the opposite effect. The main regulatory mechanism is the sensing of intracellular cholesterol in the endoplasmic reticulum by the protein SREBP (sterol regulatory element-binding protein 1 and 2). In the presence of cholesterol, SREBP is bound to two other proteins: SCAP (SREBP-cleavage-activating protein) and Insig1. When cholesterol levels fall, Insig-1 dissociates from the SREBP-SCAP complex, allowing the complex to migrate to the Golgi apparatus, where SREBP is cleaved by S1P and S2P (site-1 and -2 protease), two enzymes that are activated by SCAP when cholesterol levels are low. The cleaved SREBP then migrates to the nucleus and acts as a transcription factor to bind to the SRE (sterol regulatory element), which stimulates the transcription of many genes. Among these are the low-density lipoprotein (LDL) receptor and HMG-CoA reductase. The former scavenges circulating LDL from the bloodstream, whereas HMG-CoA reductase leads to an increase of endogenous production of cholesterol. A large part of this signaling pathway was clarified by Dr. Michael S. Brown and Dr. Joseph L. Goldstein in the 1970s. In 1985, they received the Nobel Prize in Physiology or Medicine for their work. Their subsequent work shows how the SREBP pathway regulates expression of many genes that control lipid formation and metabolism and body fuel allocation.
Cholesterol synthesis can be turned off when cholesterol levels are high, as well. HMG CoA reductase contains both a cytosolic domain (responsible for its catalytic function) and a membrane domain. The membrane domain functions to sense signals for its degradation. Increasing concentrations of cholesterol (and other sterols) cause a change in this domain's oligomerization state, which makes it more susceptible to destruction by the proteosome. This enzyme's activity can also be reduced by phosphorylation by an AMP-activated protein kinase. Because this kinase is activated by AMP, which is produced when ATP is hydrolyzed, it follows that cholesterol synthesis is halted when ATP levels are low[5].
There are two metabolic pathways of creating terpenoids:
Mevalonic acid pathway
Many organisms manufacture terpenoids through the HMG-CoA reductase pathway, the pathway that also produces cholesterol. The reactions take place in the cytosol. The pathway was discovered in the 1950s.
MEP/DOXP pathway
The 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate pathway (MEP/DOXP pathway), also known as [non-mevalonate pathway] or mevalonic acid-independent pathway, takes place in the plastids of plants and apicomplexan protozoa, as well as in many bacteria. It was discovered in the late 1980s.
Pyruvate and glyceraldehyde 3-phosphate are converted by DOXP synthase (Dxs) to 1-deoxy-D-xylulose 5-phosphate, and by DOXP reductase (Dxr, IspC) to 2-C-methyl-D-erythritol 4-phosphate (MEP). The subsequent three reaction steps catalyzed by 4-diphosphocytidyl-2-C-methyl-D-erythritol synthase (YgbP, IspD), 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (YchB, IspE), and 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (YgbB, IspF) mediate the formation of 2-C-methyl-D-erythritol 2,4-cyclopyrophosphate (MEcPP). Finally, MEcPP is converted to (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP) by HMB-PP synthase (GcpE, IspG), and HMB-PP is converted to isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) by HMB-PP reductase (LytB, IspH).
IPP and DMAPP are the end-products in either pathway, and are the precursors of isoprene, monoterpenoids (10-carbon), diterpenoids (20-carbon), carotenoids (40-carbon), chlorophylls, and plastoquinone-9 (45-carbon). Synthesis of all higher terpenoids proceeds via formation of geranyl pyrophosphate (GPP), farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate (GGPP).
Although both pathways, MVA and MEP, are mutually exclusive in most organisms, interactions between them have been reported in plants and few bacteria species.
The classical [mevalonate pathway or HMG-CoA reductase pathway is an important cellular metabolic pathway present in all higher eukaryotes and many bacteria. It is important for the production of IPP and DMAPP that serve as the basis for the biosynthesis of molecules used in processes as diverse as protein prenylation, cell membrane maintenance, hormones, protein anchoring and N-glycosylation.
Non-mevalonate pathway reactions in the biosynthesis of isoprenoids. Redrawn/transcribed verbatim from the scheme of Qidwai et al. (2014) [their Fig. 2.], see Qidwai T, Jamal F, Khan MY, Sharma B (2014). "Exploring Drug Targets in Isoprenoid Biosynthetic Pathway for Plasmodium falciparum"(review). Biochemistry Research International: 657189. doi:10.1155/2014/657189. PMC4017727. Retrieved March 23, 2017.{{cite journal}}: CS1 maint: uses authors parameter (link) Note that this source uses enzyme names and abbreviations that are both non-comprehensive and non-standard (cf. Eisenreich W, Bacher A, Arigoni D, Rohdich F (2004). "Biosynthesis of Isoprenoids Via the Non-mevalonate Pathway". Cell. Mol. Life Sci. 61 (12): 1401–26. doi:10.1007/s00018-004-3381-z. PMID15197467. Retrieved March 23, 2017.{{cite journal}}: CS1 maint: multiple names: authors list (link)); hence, image content must be reconciled with standard enzyme names and abbreviations for whichever of the pathways of the three relevant phyla/kingdoms are under discussion. See the "Non-mevalonate pathway" article at the English Wikipedia for clarification.
Eicosanoid biosynthesis begins when cell is activated by mechanical trauma, cytokines, growth factors or other stimuli.
(The stimulus may even be an eicosanoid from a neighboring cell; the pathways are complex.) This triggers the release of a phospholipase at the cell membrane.
The phospholipase travels to the nuclear membrane.
There, the phospholipase catalyzes ester hydrolysis of phospholipid (by A2) or diacylglycerol (by phospholipase C).
This frees a 20-carbon essential fatty acid. This hydrolysis appears to be the rate-determining step for eicosanoid formation.
The fatty acids may be released by any of several phospholipases.
Of these, type IV cytosolic phospholipase A2 (cPLA2) is the key actor, as cells lacking cPLA2 are generally devoid of eicosanoid synthesis. The phospholipase
cPLA2 is specific for phospholipids that contain AA, EPA or GPLA at the SN2 position.
Interestingly, cPLA2 may also release the lysophospholipid that becomes platelet-activating factor.[7]
Next, the free fatty acid is oxygenated along any of several pathways; see the Pathways table. The eicosanoid pathways (vialipoxygenase or COX) add molecular oxygen (O2). Although the fatty acid is symmetric, the resulting eicosanoids are chiral; the oxidation proceeds with high stereospecificity.
The oxidation of lipids is hazardous to cells, particularly when close to the nucleus.
There are elaborate mechanisms to prevent unwanted oxidation.
COX, the lipoxygenases and the phospholipases are tightly controlled—there are at least eight proteins activated to coordinate generation of leukotrienes.
Several of these exist in multiple isoforms.[8]
Oxidation by either COX or lipoxygenase releases reactive oxygen species (ROS) and the initial products in eicosanoid generation are themselves highly reactive peroxides.
LTA4 can form adducts with tissue DNA.
Other reactions of lipoxygenases generate cellular damage; murine models implicate 15-lipoxygenase in the pathogenesis of atherosclerosis.[9][10]
The oxidation in eicosanoid generation is compartmentalized; this limits the peroxides' damage.
The enzymes which are biosynthetic for eicosanoids (e.g. glutathione-S-transferases, epoxide hydrolases and carrier proteins) belong to families whose functions are largely involved with cellular detoxification.
This suggests that eicosanoid signaling may have evolved from the detoxification of ROS.
The cell must realize some benefit from generating lipid hydroperoxides close-by its nucleus.
PGs and LTs may signal or regulate DNA-transcription there;
LTB4 is ligand for PPARα.
(See diagram at PPAR).
Structures of Selected Eicosanoids
Prostaglandin E1. The 5-member ring is characteristic of the class.
Thromboxane A2. Oxygens have moved into the ring.
Leukotriene B4. Note the 3 conjugated double bonds.
Prostacyclin I2. The second ring distinguishes it from the prostaglandins.
Leukotriene E4, an example of a cysteinyl leukotriene.
Cyclooxygenase (COX) catalyzes the conversion of the free essential fatty acids to prostanoids by a two-step process.
In the first step, two molecules of O2 are added as two peroxide linkages and a 5-member carbon ring is forged near the middle of the fatty acid chain. This forms the short-lived, unstable intermediate Prostaglandin G (PGG).
One of the peroxide linkages sheds a single oxygen, forming PGH. (See diagrams and more detail at Cyclooxygenase).
All other prostanoids originate from PGH (as PGH1, PGH2, or PGH3).
Figure 1 shows how PGH2 (derived from Arachidonic acid) is converted:
By PGE synthetase into PGE (which in turn is converted into PGF)
The three classes of prostanoids have distinctive rings in the center of the molecule. They differ in their structures. The PGH compounds (parents to all the rest) have a 5-carbon ring, bridged by two oxygens (a peroxide.) The derived prostaglandins contain a single, unsaturated 5-carbon ring. In prostacyclins, this ring is conjoined to another oxygen-containing ring. In thromboxanes the ring becomes a 6-member ring with one oxygen.
Production of PGE2 in bacterial and viral infections appear to be stimulated by certain cytokines, e.g., interleukin-1.[7]
Leukotrienes are synthesized in the cell from arachidonic acid by 5-lipoxygenase. The catalytic mechanism involves the insertion of an oxygen moiety at a specific position in the arachidonic acid backbone.
The lipoxygenase pathway is active in leukocytes, including mast cells, eosinophils, neutrophils, monocytes, and basophils. When such cells are activated, arachidonic acid is liberated from cell membrane phospholipids by phospholipase A2, and donated by the 5-lipoxygenase-activating protein (FLAP) to 5-lipoxygenase.
5-Lipoxygenase (5-LO) uses FLAP to convert arachidonic acid into 5-hydroperoxyeicosatetraenoic acid (5-HPETE), which spontaneously reduces to 5-hydroxyeicosatetraenoic acid (5-HETE). The enzyme 5-LO acts again on 5-HETE to convert it into leukotriene A4 (LTA4), an unstable epoxide.
In cells equipped with LTA4 hydrolase, such as neutrophils and monocytes, LTA4 is converted to the dihydroxy acid leukotriene LTB4, which is a powerful chemoattractant for neutrophils acting at BLT1 and BLT2 receptors on the plasma membrane of these cells.
In cells that express LTC4 synthase, such as mast cells and eosinophils, LTA4 is conjugated with the tripeptide glutathione to form the first of the cysteinyl-leukotrienes, LTC4. Outside the cell, LTC4 can be converted by ubiquitous enzymes to form successively LTD4 and LTE4, which retain biological activity.
The cysteinyl-leukotrienes act at their cell-surface receptors CysLT1 and CysLT2 on target cells to contract bronchial and vascular smooth muscle, to increase permeability of small blood vessels, to enhance secretion of mucus in the airway and gut, and to recruit leukocytes to sites of inflammation.
Both LTB4 and the cysteinyl-leukotrienes (LTC4, LTD4, LTE4) are partly degraded in local tissues, and ultimately become inactive metabolites in the liver.
Cyclooxygenase (COX)
COX converts arachidonic acid (AA, an ω-6 PUFA) to prostaglandin H2 (PGH2), the precursor of the series-2 prostanoids. The enzyme contains two active sites: a heme with peroxidase activity, responsible for the reduction of PGG2 to PGH2, and a cyclooxygenase site, where arachidonic acid is converted into the hydroperoxy endoperoxide prostaglandin G2 (PGG2). The reaction proceeds through H atom abstraction from arachidonic acid by a tyrosine radical generated by the peroxidase active site. Two O2 molecules then react with the arachidonic acid radical, yielding PGG2.
At present, three COX isoenzymes are known: COX-1, COX-2, and COX-3. COX-3 is a splice variant of COX-1, which retains intron one and has a frameshift mutation; thus some prefer the name COX-1b or COX-1 variant (COX-1v).[3]
Different tissues express varying levels of COX-1 and COX-2. Although both enzymes act basically in the same fashion, selective inhibition can make a difference in terms of side-effects. COX-1 is considered a constitutive enzyme, being found in most mammalian cells. COX-2, on the other hand, is undetectable in most normal tissues. It is an inducible enzyme, becoming abundant in activated macrophages and other cells at sites of inflammation. More recently, it has been shown to be upregulated in various carcinomas and to have a central role in tumorigenesis.
Both COX-1 and -2 (also known as PGHS-1 and -2) also oxygenate two other essential fatty acids – DGLA (ω-6) and EPA (ω-3) – to give the series-1 and series-3 prostanoids, which are less inflammatory than those of series-2. DGLA and EPA are competitive inhibitors with AA for the COX pathways. This inhibition is a major mode of action in the way that dietary sources of DGLA and EPA (e.g., borage, fish oil) reduce inflammation.The use of anabolic steroids is now banned by all major sporting bodies, including the ATP, WTA, ITF, International Olympic Committee, FIFA, UEFA, all major professional golf tours, the National Hockey League, Major League Baseball, the National Basketball Association, the European Athletic Association, WWE and the NFL. However drug testing can be wildly inconsistent and, in some instances, has gone unenforced.
Bile acids are steroid acids found predominantly in the bile of mammals. Bile salts are bile acids compounded with a cation, usually sodium. In humans, the salts of taurocholic acid and glycocholic acid (derivatives of cholic acid) represent approximately eighty percent of all bile salts. The two major bile acids are cholic acid, and chenodeoxycholic acid. Bile acids, glycine and taurine conjugates, and 7-alpha-dehydroxylated derivatives (deoxycholic acid and lithocholic acid) are all found in human intestinal bile. An increase in bile flow is exhibited with an increased secretion of bile acids. The main function of bile acid is to facilitate the formation of micelles, which promotes processing of dietary fat.
Bile acids are made in the liver by the cytochrome P450-mediated oxidation of cholesterol. They are conjugated with taurine or the amino acid glycine, or with a sulfate or a glucuronide, and are then stored in the gallbladder, which concentrates the salts by removing the water. In humans, the rate limiting step is the addition of a hydroxyl group on position 7 of the steroid nucleus by the enzyme cholesterol 7 alpha-hydroxylase. Upon eating a meal, the contents of the gallbladder are secreted into the intestine, where bile acids serve the purpose of emulsifying dietary fats. Bile acids serve other functions, including eliminating cholesterol from the body, driving the flow of bile to eliminate catabolites from the liver, emulsifying lipids and fat soluble vitamins in the intestine to form micelles that can be transported via the lacteal system, and aiding in the reduction of the bacteria flora found in the small intestine and biliary tract.
Bile acid refers to the protonated (-COOH) form. Bile salt refers to the deprotonated or ionized (-COO-) form. Conjugated bile acids are more efficient at emulsifying fats because at intestinal pH, they are more ionized than unconjugated bile acids.[11]
Synthesis of bile acids is a major route of cholesterol metabolism in most species other than humans. The body produces about 800 mg of cholesterol per day and about half of that is used for bile acid synthesis. In total about 20-30 grams of bile acids are secreted into the intestine daily. About 90% of excreted bile acids are reabsorbed by active transport in the ileum and recycled in what is referred to as the enterohepatic circulation which moves the bile salts from the intestinal system back to the liver and the gallbladder. This allows a low rate of daily synthesis, but high secretion to the digestive system. Bile is also used to break down fat globules into tiny droplets. Bile from slaughtered animals can be used in the preparation of soap.
Glycolipids
The heads of glycolipids contain a sphingosine with one or several sugar units attached to it. The hydrophobic chains belong either to:
two fatty acids - in the case of the phosphoglycerides.
one FA and the hydrocarbon tail of sphingosine - in the case of sphingomyelin and the glycolipids.
Fatty acids
The fatty acids in phospho- and glycolipids usually contain an even number of carbon atoms, typically between 14 and 24. The 16- and 18-carbon FAs are the most common ones. FAs may be saturated or unsaturated, with the configuration of the double bonds nearly always cis. The length and the degree of unsaturation of FAs chains have a profound effect on membranes' fluidity.
Phosphoglycerides
In phosphoglycerides, the hydroxyl groups at C-1 and C-2 of glycerol are esterified to the carboxyl groups of the FAs. The C-3 hydroxyl group is esterified to phosphoric acid. The resulting compound, called phosphatidate, is the simplest phosphoglycerate. Only small amounts of phosphatidate are present in membranes. However, it is a key intermediate in the biosynthesis of the other phosphoglycerides.
Sphingosine
Sphingosine is an amino alcohol that contains a long, unsaturated hydrocarbon chain. In sphingomyelin and glycolipids, the amino group of sphingosine is linked to FAs by an amide bond. In sphingomyelin the primary hydroxyl group of sphingosine is esterified to phosphoryl choline. In glycolipids, the sugar component is attached to this group. The simplest glycolipid is cerebroside, in which there is only one sugar residue, either Glc or Gal. More complex glycolipids, such as gangliosides, contain a branched chain of as many as seven sugar residues.
Sphingosine is synthesized from palmitoyl CoA and serine in a condensation required to yield dehydrosphingosine.
Sphingosine is synthesized from palmitoyl CoA and serine in a condensation required to yield dehydrosphingosine
Dehydrosphingosine is then reduced by NADPH to dihydrosphingosine (sphinganine), and finally oxidized by FAD to sphingosine.
There is no direct route of synthesis from sphinganine to sphingosine; it has to be acylated first to dihydroceramide, which is then dehydrogenated to ceramide. Sphingosine is formed via degradation of sphingolipid in the lysosome.
Cholesterol
Cholesterol occurs naturally in eukaryotecell membranes where it is bio-synthesised from mevalonate via a squalene cyclisation of terpenoids. It associated preferentially with sphingolipids (see diagram) in cholesterol-rich lipid rafts areas of the membranes in eukaryotic cells.[12] Hopanoids serve a similar function in prokaryotes.
Cell membranes require high levels - typically an average of 20% cholesterol molecular in the whole membrane, increasing locally in raft areas up to 50% cholesterol (- % is molecular ratio) [13] Formation of lipid rafts promotes aggregation of peripheral and transmembrane proteins including docking of SNARE and VAMP proteins[14]
Membrane structures. Top, an archaeal phospholipid: 1, isoprene chains; 2, ether linkages; 3, L-glycerol moiety; 4, phosphate group. Middle, a bacterial or eukaryotic phospholipid: 5, fatty acid chains; 6, ester linkages; 7, D-glycerol moiety; 8, phosphate group. Bottom: 9, lipid bilayer of bacteria and eukaryotes; 10, lipid monolayer of some archaea.
Archaeal membranes are made of molecules that differ strongly from those in other life forms, showing that archaea are related only distantly to bacteria and eukaryotes.[15] In all organisms cell membranes are made of molecules known as phospholipids. These molecules possess both a polar part that dissolves in water (the phosphate "head"), and a "greasy" non-polar part that does not (the lipid tail). These dissimilar parts are connected by a glycerol moiety. In water, phospholipids cluster, with the heads facing the water and the tails facing away from it. The major structure in cell membranes is a double layer of these phospholipids, which is called a lipid bilayer[16].
These phospholipids are unusual in four ways:
Bacteria and eukaryotes have membranes composed mainly of glycerol-esterlipids, whereas archaea have membranes composed of glycerol-ether lipids.[17] The difference is the type of bond that joins the lipids to the glycerol moiety; the two types are shown in yellow in the figure at the right. In ester lipids this is an ester bond, whereas in ether lipids this is an ether bond. Ether bonds are chemically more resistant than ester bonds. This stability might help archaea to survive extreme temperatures and very acidic or alkaline environments.[18] Bacteria and eukaryotes do contain some ether lipids, but in contrast to archaea these lipids are not a major part of their membranes.
The stereochemistry of the glycerol moiety is the reverse of that found in other organisms. The glycerol moiety can occur in two forms that are mirror images of one another, called the right-handed and left-handed forms; in chemistry these are called enantiomers. Just as a right hand does not fit easily into a left-handed glove, a right-handed glycerol molecule generally cannot be used or made by enzymes adapted for the left-handed form. This suggests that archaea use entirely different enzymes for synthesizing phospholipids than do bacteria and eukaryotes. Such enzymes developed very early in life's history, suggesting an early split from the other two domains.[15]
Archaeal lipid tails are chemically different from other organisms. Archaeal lipids are based upon the isoprenoid sidechain and are long chains with multiple side-branches and sometimes even cyclopropane or cyclohexane rings.[19] This is in contrast to the fatty acids found in other organisms' membranes, which have straight chains with no branches or rings. Although isoprenoids play an important role in the biochemistry of many organisms, only the archaea use them to make phospholipids. These branched chains may help prevent archaean membranes from leaking at high temperatures.[20]
In some archaea the lipid bilayer is replaced by a monolayer. In effect, the archaea fuse the tails of two independent phospholipid molecules into a single molecule with two polar heads; this fusion may make their membranes more rigid and better able to resist harsh environments.[21] For example, the lipids in Ferroplasma are of this type, which is thought to aid this organism's survival in its highly acidic habitat.[22]
Terpenes are derived biosynthetically from units of isoprene, which has the molecular formula C5H8. The basic molecular formulae of terpenes are multiples of that, (C5H8)n where n is the number of linked isoprene units. This is called the isoprene rule or the C5 rule. The isoprene units may be linked together "head to tail" to form linear chains or they may be arranged to form rings. One can consider the isoprene unit as one of nature's common building blocks.
Isoprene itself does not undergo the building process, but rather activated forms, isopentenyl pyrophosphate (IPP or also isopentenyl diphosphate) and dimethylallyl pyrophosphate (DMAPP or also dimethylallyl diphosphate), are the components in the biosynthetic pathway. IPP is formed from acetyl-CoA via the intermediacy of mevalonic acid in the HMG-CoA reductase pathway. An alternative, totally unrelated biosynthesis pathway of IPP is known in some bacterial groups and the plastids of plants, the so-called MEP(2-Methyl-D-erythritol-4-phosphate)-pathway, which is initiated from C5-sugars. In both pathways, IPP is isomerized to DMAPP by the enzyme isopentenyl pyrophosphate isomerase.
Dimethylallyl pyrophosphate
Isopentenyl pyrophosphate
As chains of isoprene units are built up, the resulting terpenes are classified sequentially by size as hemiterpenes, monoterpenes, sesquiterpenes, diterpenes, sesterterpenes, triterpenes, and tetraterpenes[23].
Several key enzymes can be activated through DNA transcription al regulation on activation of SREBP (sterol regulatory element-binding protein-1 and -2). This intracellular sensor detects low cholesterol levels and stimulates endogenous production by the HMG-CoA reductase pathway, as well as increasing lipoprotein uptake by up-regulating the LDL-receptor. Regulation of this pathway is also achieved by controlling the rate of translation of the mRNA, degradation of reductase and phosphorylation.
HMG-CoA is reduced to mevalonate by NADPH. This reaction occurs in the cytosol. It is the rate limiting step in cholesterol synthesis, which is why the enzyme catalyzing the reaction is a target of statins.
The HMG-CoA reductase pathway, which is blocked by statins via inhibiting the rate limiting enzyme HMG-CoA reductase.
In 1971, Akira Endo, a Japanese biochemist working for the drug company Sankyo, began the search for a cholesterol-lowering drug. Research had already shown that cholesterol is mostly manufactured by the body in the liver, using an enzyme known as HMG-CoA reductase. Endo and his team reasoned that certain microorganisms may produce inhibitors of the enzyme to defend themselves against other organisms, as mevalonate is a precursor of many substances required by organisms for the maintenance of their cell wall (ergosterol) or cytoskeleton (isoprenoids). The first agent they identified was mevastatin (ML-236B), a molecule produced by the fungus Penicillium citrinum.
Journalist John Simons writes in Fortune magazine that word of the discovery spread quickly through the medical community, as did rumors linking the statin to tumors, muscle deterioration, and sometimes death in laboratory dogs. Several drug companies were put off by these reports, but P. Roy Vagelos, the chief scientist and later CEO of Merck & Co, showed an interest, and made several trips to Japan starting in 1975. By 1978, Merck had isolated lovastatin (mevinolin, MK803) from the fungus Aspergillus terreus, first marketed in 1987 as Mevacor.
A link between cholesterol and cardiovascular disease, known as the lipid hypothesis, had already been suggested. Cholesterol is the main constituent of atheroma, the fatty lumps in the wall of arteries that occur in atherosclerosis and, when ruptured, cause the vast majority of heart attacks. Treatment consisted mainly of dietary measures such as a low-fat diet, and poorly tolerated medicines such as clofibrate, cholestyramine and nicotinic acid. Cholesterol researcher Daniel Steinberg writes that while the Coronary Primary Prevention Trial of 1984 demonstrated that cholesterol lowering could significantly reduce the risk of heart attacks and angina, physicians, including cardiologists, remained largely unconvinced.
To market statins effectively, Merck had to convince the public about the dangers of high cholesterol, and doctors that statins were safe and would extend lives. As a result of public campaigns, people became familiar with their cholesterol numbers and the difference between "good" and "bad" cholesterol, and rival pharmaceutical companies began producing their own statins, such as pravastatin (Pravachol), manufactured by Sankyo and Bristol-Myers Squibb. In April 1994, the results of a Merck-sponsored study, the Scandinavian Simvastatin Survival Study or "4S", were announced. Researchers tested simvastatin, later sold by Merck as Zocor, on 4,444 patients with high cholesterol and heart disease. After five years, the study concluded that patients saw a 35-percent reduction in their cholesterol, and their chances of dying of a heart attack were reduced by 42 percent. In 1995, Zocor and Mevacor both made Merck over US$1 billion. Endo was awarded the 2006 Japan Prize, and the Lasker-DeBakey Clinical Medical Research Award in 2008.
Statins act by competitively inhibiting HMG-CoA reductase, the first committed enzyme of the HMG-CoA reductase pathway. Because statins are similar to HMG-CoA on a molecular level they take the place of HMG-CoA in the enzyme and reduce the rate by which it is able to produce mevalonate, the next molecule in the cascade that eventually produces cholesterol, as well as a number of other compounds. This ultimately reduces cholesterol via several mechanisms[25].
Inhibiting cholesterol synthesis
By inhibiting HMG-CoA reductase, statins block the pathway for synthesizing cholesterol in the liver. This is significant because most circulating cholesterol comes from internal manufacture rather than the diet. When the liver can no longer produce cholesterol, levels of cholesterol in the blood will fall. Cholesterol synthesis appears to occur mostly at night, so statins with short half-lives are usually taken at night to maximize their effect. Studies have shown greater LDL and total cholesterol reductions in the short-acting simvastatin taken at night rather than the morning, but have shown no difference in the long-acting atorvastatin.
Increasing LDL uptake
Liver cells sense the reduced levels of liver cholesterol and seek to compensate by synthesizing LDL receptors to draw cholesterol out of the circulation. This is accomplished via protease enzymes that cleave a protein called "membrane-bound sterol regulatory element binding protein", which migrates to the nucleus and causes increased production of various other proteins and enzymes, including the LDL receptor. The LDL receptor then relocates to the liver cell membrane and binds to passing LDL and VLDL particles (the "bad cholesterol" linked to disease). LDL and VLDL are drawn out of circulation into the liver where the cholesterol is reprocessed into bile salts. These are excreted, and subsequently recycled mostly by an internal bile salt circulation.
Other effects
Statins exhibit action beyond lipid-lowering activity in the prevention of atherosclerosis. The ASTEROID trial showed direct ultrasound evidence of atheroma regression during statin therapy. Researchers hypothesize that statins prevent cardiovascular disease via four proposed mechanisms (all subjects of a large body of biomedical research):
Improve endothelial function,
Modulate inflammatory responses,
Maintain plaque stability,
Prevent thrombus formation,
Statins may even benefit those without high cholesterol. In 2008 the JUPITER study showed fewer stroke, heart attacks, and surgeries even for patients who had no history of high cholesterol or heart disease, but only elevated C-reactive protein levels. There were also 20% fewer deaths (mainly from reduction in cancer deaths) though deaths from cardiovascular causes were not reduced
Adverse effect of statin
The most common adverse side effects are raised liver enzymes and muscle problems. In randomized clinical trials, reported adverse effects are low; but they are "higher in studies of real world use", and more varied. In randomized trials statins increased the risk of an adverse effect by 39% compared to placebo (odds ratios 1.4); two-thirds of these were myalgia or raised liver enzymes with serious adverse effects similar to placebo. However, reliance on clinical trials can be misleading indications of real-world adverse effects – for example, the statin cerivastatin was withdrawn from the market in 2001 due to cases of rhabdomyolysis (muscle breakdown), although rhabdomyolysis did not occur in a meta-analysis of cerivastatin clinical trials. Other possible adverse effects include cognitive loss, neuropathy, pancreatic and hepatic dysfunction, and sexual dysfunction.
Some patients on statin therapy report myalgias, muscle cramps,] or, less frequently, gastrointestinal or other symptoms. Liver enzyme derangements may also occur, typically in about 0.5%, are also seen at similar rates with placebo use and repeated enzyme testing, and generally return to normal either without discontinuance over time or after briefly discontinuing the drug. Multiple other side-effects occur rarely; typically also at similar rates with only placebo in the large statin safety/efficacy trials. Two randomized clinical trials found cognitive issues while two did not; recurrence upon reintroduction suggests that these are causally related to statins in some individuals. A Danish case-control study published in 2002 suggested a relation between long term statin use and increased risk of nerve damage or polyneuropathy but suggested this side effect is "rare, but it does occur"; other researchers have pointed to studies of the effectiveness of statins in trials involving 50,000 people which have not shown nerve damage as a significant side effect[26].
Acetyl-CoA carboxylase (ACC):
Prokaryotes and plants have multi-subunit ACCs composed of several polypeptides encoded by distinct genes. Biotin carboxylase (BC) activity, biotin carboxyl carrier protein (BCCP), and carboxyl transferase (CT) activity are each contained on a different subunit. The stoichiometry of these subunits in the ACC holoenzyme differs amongst organisms. Humans and most eukaryotes have evolved an ACC with CT and BC catalytic domains and biotin carboxyl carrier domains on a single polypeptide. ACC functional regions, starting from the N-terminus to C-terminus are the biotin carboxylase (BC), biotin binding (BB), carboxyltransferase (CT), and ATP-binding (AB). AB lies within BC. Biotin is covalently attached through an amide bond to the long side chain of a lysine reside in BB. As BB is between BC and CT regions, biotin can be easily translocate to both of the active sites where it is required.
In mammals where two isoforms of ACC are expressed, the main structural difference between these isoforms is the extended ACC2 N-terminus containing a mitochondria targeting sequence.
Crystallographic structures of E. Coli Acetyl CoA Carboxylase
Cartoon diagram of Biotin Carboxylase of E.Coli Acetyl CoA Carboxylase
Biotin Carboxyl Carrier Protein of E. Coli Acetyl CoA Carboxylase
Carboxyltransferase Subunit of E. Coli Acetyl CoA Carboxylase
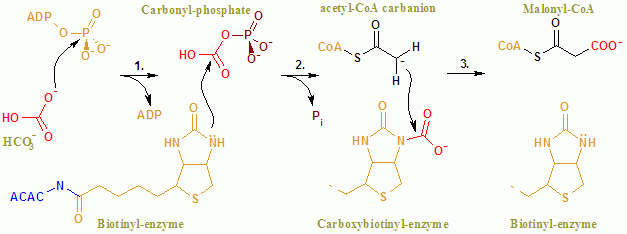
The overall reaction of ACAC(A,B) proceeds by a two-step mechanism.[27] The first reaction is carried out by BC and involves the ATP-dependent carboxylation of biotin with bicarbonate serving as the source of CO2. The carboxyl group is transferred from biotin to acetyl CoA to form malonyl CoA in the second reaction, which is catalyzed by CT.
The reaction mechanism of ACAC(A,B).
The color scheme is as follows: enzyme, coenzymes, substrate names, metal ions, phosphate, and carbonate
In the context of the active site, the reaction proceeds with extensive interaction of the residues Glu296 and positively charged Arg338 and Arg292 with the substrates.[28] Two Mg2+ are coordinated by the phosphate groups on the ATP, and are required for ATP binding to the enzyme. Bicarbonate is deprotonated by Glu296, although in solution, this proton transfer is unlikely as the pKa of bicarbonate is 10.3. The enzyme apparently manipulates pKas to facilitate the deprotonation of bicarbonate. The pKa of bicarbonate is decreased by its interaction with positively charged side chains of Arg338 and Arg292. Furthermore, Glu296 interacts with the side chain of Glu211, an interaction that has been shown to cause an increase in the apparent pKa. Following deprotonation of bicarbonate, the oxygen of the bicarbonate acts as a nucleophile and attacks the gamma phosphate on ATP. The carboxyphosphate intermediate quickly decomposes to CO2 and PO43-. The PO43- deprotonates biotin, creating an enolate, stabilized by Arg338, that subsequently attacks [[CO2]] resulting in the production of carboxybiotin.[28] The carboxybiotin translocates to the carboxytransferase (CT) active site, where the carboxyl group is transferred to acetyl-CoA. In contrast to the BC domain, little is known about the reaction mechanism of CT. A proposed mechanism is the release of carbon dioxide from biotin, which subsequently abstracts a proton from the methyl group from acetyl CoA carboxylase. The resulting enolate attacks CO2 to form malonyl CoA. In a competing mechanism, proton abstraction is concerted with the attack of acetyl CoA.The function of ACAC is to regulate the metabolism of fatty acids. When the enzyme is active, the product, malonyl-CoA is produced which is a building block for new fatty acids and can inhibit the transfer of the fatty acyl group from acyl CoA to carnitine with carnitine acyltransferase, which inhibits the beta-oxidation of fatty acids in the mitochondria.
In mammals, two main isoforms of ACC are expressed, ACC1 and ACC2, which differ in both tissue distribution and function. ACC1 is found in the cytoplasm of all cells but is enriched in lipogenic tissue, such as adipose tissue and lactating mammary glands, where fatty acid synthesis is important. In oxidative tissues, such as the skeletal muscle and the heart, the ratio of ACC2 expressed is higher. ACC1 and ACC2 are both highly expressed in the liver where both fatty acid oxidation and synthesis is important. The differences in tissue distribution indicate that ACC1 maintains regulation of fatty acid synthesis whereas ACC2 mainly regulates fatty acid oxidation.
The classical mevalonate pathway or HMG-CoA reductase pathway is an important cellular metabolic pathway present in all higher eukaryotes and many bacteria. It is important for the production of IPP and DMAPP that serve as the basis for the biosynthesis of molecules used in processes as diverse as protein prenylation, cell membrane maintenance, hormones, protein anchoring and N-glycosylation.
In contrast to the classical mevalonate pathway of isoprenoid biosynthesis, plants and apicomplexan protozoa such as malaria parasites have the ability to produce their isoprenoids (terpenoids) using an alternative pathway, the non-mevalonate pathway, which takes place in their plastids.[29] In addition, most bacteria including important pathogens such as Mycobacterium tuberculosis synthesize IPP and DMAPP via the non-mevalonate pathway.
Non-mevalonate pathway reactions in the biosynthesis of isoprenoids. Redrawn/transcribed verbatim from the scheme of Qidwai et al. (2014) [their Fig. 2.], see Qidwai T, Jamal F, Khan MY, Sharma B (2014). "Exploring Drug Targets in Isoprenoid Biosynthetic Pathway for Plasmodium falciparum"(review). Biochemistry Research International: 657189. doi:10.1155/2014/657189. PMC4017727. Retrieved March 23, 2017.{{cite journal}}: CS1 maint: uses authors parameter (link) Note that this source uses enzyme names and abbreviations that are both non-comprehensive and non-standard (cf. Eisenreich W, Bacher A, Arigoni D, Rohdich F (2004). "Biosynthesis of Isoprenoids Via the Non-mevalonate Pathway". Cell. Mol. Life Sci. 61 (12): 1401–26. doi:10.1007/s00018-004-3381-z. PMID15197467. Retrieved March 23, 2017.{{cite journal}}: CS1 maint: multiple names: authors list (link)); hence, image content must be reconciled with standard enzyme names and abbreviations for whichever of the pathways of the three relevant phyla/kingdoms are under discussion. See the "Non-mevalonate pathway" article at the English Wikipedia for clarification.
Fosmidomycin is an antibiotic that specifically inhibits DXP reductoisomerase, a key enzyme in the non-mevalonate pathway of isoprenoid biosynthesis. It is a structural analogue of 2-C-methyl-D-erythrose 4-phosphate. It inhibits the E. coli enzyme with a KI value of 38 nM (4), MTB at 80 nM, and the Francisella enzyme at 99 nM.
The discovery of the non-mevalonate pathway in malaria parasites has indicated the use of fosmidomycin and other such inhibitors as antimalarial drugs. Indeed, fosmidomycin has been tested in combination treatment with clindamycin for treatment of malaria with favorable results. It has been shown that an increase in copy number of the target enzyme (DXP reductoisomerase) correlates with in vitro fosmidomycin resistance in the lethal malaria parasite, Plasmodium falciparum.
↑Lichtenthaler H (1999). "The 1-Deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants". Annu Rev Plant Physiol Plant Mol Biol. 50: 47–65. doi:10.1146/annurev.arplant.50.1.47. PMID15012203.
↑ abUniversity of Kansas Medical Center (2004). "Eicosanoids and Inflammation"(PDF). Retrieved 2007-01-05.
[dead link]Invalid <ref> tag; name "University" defined multiple times with different content
↑Schewe T. (2002 Mar-Apr). "15-lipoxygenase-1: a prooxidant enzyme". Biol Chem. 383 (3–4): 365–74. doi:10.1515/BC.2002.041. PMID12033428. {{cite journal}}: |access-date= requires |url= (help); Check date values in: |year= (help)
↑'Essentials of Medical Biochemistry, Lieberman, Marks and Smith, eds, p432, 2007'
↑
Chen, H;Born, E;Mathur, S N;Field, F J, J Lipid, Res Dec12 2159-67
Cholesterol and sphingomyelin syntheses are regulated independently in cultured human intestinal cells, CaCo-2: role of membrane cholesterol and sphingomyelin content
¦Volume= 34
¦Year= 1993
¦PMID=8301234
¦ISBN=0022-2275
↑de Meyer F, Smit B. Effect of cholesterol on the structure of a phospholipid bilayer. Proc Natl
Acad Sci U S A 2009; 106: 3654-8.
↑Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele C, Jahn R. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis EMBO J 2001;20:2202-13.
↑Lee CK, Cheong HK, Ryu KS, Lee JI, Lee W, Jeon YH, Cheong C (August 2008). "Biotinoyl domain of human acetyl-CoA carboxylase: Structural insights into the carboxyl transfer mechanism". Proteins. 72 (2): 613–24. doi:10.1002/prot.21952. PMID18247344.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑Lichtenthaler H (1999). "The 1-Deoxy-D-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants". Annu Rev Plant Physiol Plant Mol Biol. 50: 47–65. doi:10.1146/annurev.arplant.50.1.47. PMID15012203.
↑Eisenreich W, Bacher A, Arigoni D, Rohdich F. Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci. 2004;61:1401-1426. PMID 15197467










 Sphingosine - IUPAC Name: (2S,3R)-2-aminooctadec-4-ene-1,3-diol
Sphingosine - IUPAC Name: (2S,3R)-2-aminooctadec-4-ene-1,3-diol Sphingosine is synthesized from palmitoyl CoA and serine in a condensation required to yield dehydrosphingosine
Sphingosine is synthesized from palmitoyl CoA and serine in a condensation required to yield dehydrosphingosine













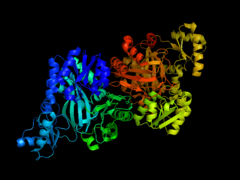 Cartoon diagram of Biotin Carboxylase of E.Coli Acetyl CoA Carboxylase
Cartoon diagram of Biotin Carboxylase of E.Coli Acetyl CoA Carboxylase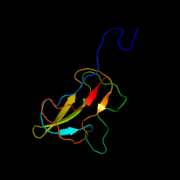 Biotin Carboxyl Carrier Protein of E. Coli Acetyl CoA Carboxylase
Biotin Carboxyl Carrier Protein of E. Coli Acetyl CoA Carboxylase Carboxyltransferase Subunit of E. Coli Acetyl CoA Carboxylase
Carboxyltransferase Subunit of E. Coli Acetyl CoA Carboxylase