Gluconeogenesis pathway with key molecules and enzymes. Many steps are the opposite of those found in the glycolysis.
Gluconeogenesis (abbreviated GNG) is a metabolic pathway that results in the generation of glucose from non-carbohydrate carbon substrates such as lactate, glycerol, and glucogenic amino acids.
It is one of the two main mechanisms humans and many other animals use to keep blood glucose levels from dropping too low (hypoglycemia). The other means of maintaining blood glucose levels is through the degradation of glycogen (glycogenolysis).
Gluconeogenesis is a ubiquitous process, present in plants, animals, fungi, bacteria, and other microorganisms. In animals, gluconeogenesis takes place mainly in the liver and, to a lesser extent, in the cortex of kidneys. This process occurs during periods of fasting, starvation, low-carbohydrate diets, or intense exercise and is highly endergonic. For example, the pathway leading from phosphoenolpyruvate to glucose-6-phosphate requires 6 molecules of ATP. Gluconeogenesis is often associated with ketosis. Gluconeogenesis is also a target of therapy for type II diabetes, such as metformin, which inhibits glucose formation and stimulates glucose uptake by cells.
Lactate is transported back to the liver where it is converted into pyruvate by the Cori cycle using the enzyme lactate dehydrogenase. Pyruvate, the first designated substrate of the gluconeogenic pathway, can then be used to generate glucose. All citric acid cycle intermediates, through conversion to oxaloacetate, amino acids other than lysine or leucine, and glycerol can also function as substrates for gluconeogenesis.Transamination or deamination of amino acids facilitates entering of their carbon skeleton into the cycle directly (as pyruvate or oxaloacetate), or indirectly via the citric acid cycle.
Whether fatty acids can be converted into glucose in animals has been a longstanding question in biochemistry. It is known that odd-chain fatty acids can be oxidized to yield propionyl CoA, a precursor for succinyl CoA, which can be converted to pyruvate and enter into gluconeogenesis. In plants, to be specific, in seedlings, the glyoxylate cycle can be used to convert fatty acids (acetate) into the primary carbon source of the organism. The glyoxylate cycle produces four-carbon dicarboxylic acids that can enter gluconeogenesis.
In 1995, researchers identified the glyoxylate cycle in nematodes. In addition, the glyoxylate enzymes malate synthase and isocitrate lyase have been found in animal tissues. Genes coding for malate synthase gene have been identified in other [metazoans] including arthropods, echinoderms, and even some vertebrates. Mammals found to possess these genes include monotremes (platypus) and marsupials (opossum) but not placental mammals. Genes for isocitrate lyase are found only in nematodes, in which, it is apparent, they originated in horizontal gene transfer from bacteria.
The existence of glyoxylate cycles in humans has not been established, and it is widely held that fatty acids cannot be converted to glucose in humans directly. However, carbon-14 has been shown to end up in glucose when it is supplied in fatty acids. Despite these findings, it is considered unlikely that the 2-carbon acetyl-CoA derived from the oxidation of fatty acids would produce a net yield of glucose via the citric acid cycle. However, it is possible that, with additional sources of carbon via other pathways, glucose could be synthesized from acetyl-CoA. In fact, it is known that Ketone bodies, β-hydroxybutyrate in particular, can be converted to glucose at least in small amounts (β-hydroxybutyrate to acetoacetate to acetone to propanediol to pyruvate to glucose).
Glycerol, which is a part of the triacylglycerol molecule, can be used in gluconeogenesis.
In humans, gluconeogenesis is restricted to the liver and to a lesser extent the kidney.
In all species, the formation of oxaloacetate from pyruvate and TCA cycle intermediates is restricted to the mitochondrion, and the enzymes that convert PEP to glucose are found in the cytosol. The location of the enzyme that links these two parts of gluconeogenesis by converting oxaloacetate to PEP, PEP carboxykinase, is variable by species: it can be found entirely within the mitochondria, entirely within the cytosol, or dispersed evenly between the two, as it is in humans. Transport of PEP across the mitochondrial membrane is accomplished by dedicated transport proteins; however no such proteins exist for oxaloacetate. Therefore species that lack intra-mitochondrial PEP, oxaloacetate must be converted into malate or asparate, exported from the mitochondrion, and converted back into oxaloacetate in order to allow gluconeogenesis to continue.
Gluconeogenesis is a pathway consisting of eleven enzyme-catalyzed reactions. The pathway can begin in the mitochondria or cytoplasm, depending on the substrate being used. Many of the reactions are the reversible steps found in glycolysis.
Gluconeogenesis begins in the mitochondria with the formation of oxaloacetate through carboxylation of pyruvate. This reaction also requires one molecule of ATP, and is catalyzed by pyruvate carboxylase. This enzyme is stimulated by high levels of acetyl-CoA (produced in β-oxidation in the liver) and inhibited by high levels of ADP.
Oxaloacetate is reduced to malate using NADH, a step required for transport out of the mitochondria.
Malate is oxidized to oxaloacetate using NAD+ in the cytoplasm, where the remaining steps of gluconeogenesis occur.
Oxaloacetate is decarboxylated and phosphorylated to produce phosphoenolpyruvate by phosphoenolpyruvate carboxykinase. One molecule of GTP is hydrolyzed to GDP during this reaction.
The next steps in the reaction are the same as reversed glycolysis. However, fructose-1,6-bisphosphatase converts fructose-1,6-bisphosphate to fructose 6-phosphate, requiring one water molecule and releasing one phosphate. This is also the rate-limiting step of gluconeogenesis.
Glucose-6-phosphate is formed from fructose 6-phosphate by phosphoglucoisomerase. Glucose-6-phosphate can be used in other metabolic pathways or dephosphorylated to free glucose. Whereas free glucose can easily diffuse in and out of the cell, the phosphorylated form (glucose-6-phosphate) is locked in the cell, a mechanism by which intracellular glucose levels are controlled by cells.
The final reaction of gluconeogenesis, the formation of glucose, occurs in the lumen of the endoplasmic reticulum, where glucose-6-phosphate is hydrolyzed by glucose-6-phosphatase to produce glucose. Glucose is shuttled into the cytosol by glucose transporters located in the membrane of the endoplasmic reticulum.
While most steps in gluconeogenesis are the reverse of those found in glycolysis, three regulated and strongly exergonic reactions are replaced with more kinetically favorable reactions. Hexokinase/glucokinase, phosphofructokinase, and pyruvate kinase enzymes of glycolysis are replaced with glucose-6-phosphatase, fructose-1,6-bisphosphatase, and PEP carboxykinase. This system of reciprocal control allow glycolysis and gluconeogenesis to inhibit each other and prevent the formation of a futile cycle.
The majority of the enzymes responsible for gluconeogenesis are found in the cytoplasm; the exceptions are mitochondrial pyruvate carboxylase and, in animals, phosphoenolpyruvate carboxykinase. The latter exists as an isozyme located in both the mitochondrion and the cytosol. The rate of gluconeogenesis is ultimately controlled by the action of a key enzyme, fructose-1,6-bisphosphatase, which is also regulated through signal tranduction by cAMP and its phosphorylation.
Most factors that regulate the activity of the gluconeogenesis pathway do so by inhibiting the activity or expression of key enzymes. However, both acetyl CoA and citrate activate gluconeogenesis enzymes (pyruvate carboxylase and fructose-1,6-bisphosphatase, respectively). Due to the reciprocal control of the cycle, acetyl-CoA and citrate also have inhibitory roles in the activity of pyruvate kinase.
During gluconeogenesis, pyruvate carboxylase is the first enzyme in the pathway that synthesizes phosphoenolpyruvate (PEP) from pyruvate. The enzyme pyruvate carboxylase acts within the mitochodrial matrix to convert pyruvate to oxaloacetate (OAA), utilizing the energy from the hydrolysis of one molecule of ATP.
In the next step, OAA is then decarboxylated and simultaneously phosphorylated, which is catalyzed by one of two isoforms of phosphoenolpyruvate carboxykinase (PEPCK) either in the cytosol or in the mitochondria to produce PEP. Under ordinary gluconeogenic condition, OAA is converted into PEP by mitochondrial PEPCK; the resultant PEP is then transported out of the mitochondria via the citric acid cycle carrier system, and converted into glucose by cytosolic gluconeogenic enzymes. However, during starvation when cytostolic NADH concentration is low and mitochrondrial NADH levels are high, oxaloacetate can be used as a shuttle of reducing equivalents. As such OAA is converted into malate by mitochondrial malate dehydrogenase (MDH). After export into the cytosol, malate is converted back into OAA, with contaminant reduction of NAD+; OAA is subsequently converted to PEP which is available for gluconeogenesis in the cytosol along with the transported reducing equivalent NADH.
Very high levels of PC activity, together with high activities of other gluconeogenic enzymes including PEPCK, fructose-1,6-bisphosphatase and glucose-6-phosphatase in liver and kidney cortex, suggest that a primary role of PC is to participate in gluconeogenesis in these organs. During fasting or starvation when endogenous glucose is required for certain tissues (brain, white blood cells and kidney medulla), expression of PC and other gluconeogenic enzymes is elevated. In rats and mice, alteration of nutrition status has been shown to affect hepatic PC activity. Fasting promotes hepatic glucose production sustained by an increased pyruvate flux, and increases in PC activity and protein concentration; Diabetes similarly increases gluconeogenesis through enhanced uptake of substrate and increased flux through liver PC in mice and rats Similarly to other gluconeogenic enzymes, PC is positively regulated by glucagon and glucocorticoids while negatively regulated by insulin. Further supporting the key role of PC in gluconeogenesis, in dairy cattle, which have hexose absorption ability at adequate nutrition levels, PC and the associated gluconeogenic enzyme PEPCK are markedly elevated during the transition to lactation in proposed support of lactose synthesis for milk production.
Aside from the role of PC in gluconeogenesis, PC serves an anaplerotic role (an enzyme catalyzed reaction that can replenish the supply of intermediates in the citric acid cycle) for the tricarboxylic acid cycle (essential to provide oxaloacetate), when intermediates are removed for different biosynthetic purposes.
Phosphoenolpyruvate carboxykinase (PEPCK) is an enzyme in the lyase family used in the metabolic pathway of gluconeogenesis. It converts oxaloacetate into phosphoenolpyruvate and carbon dioxide.
It is found in two forms, cytosolic and mitochondrial.
It has been shown that PEPCK catalyzes the rate-controlling step of gluconeogenesis, the process whereby glucose is synthesized. The enzyme has therefore been thought to be essential in glucose homeostasis, as evidenced by laboratory mice that contracted diabetes mellitus type 2 as a result of the overexpression of PEPCK.
A recent study suggests that the role that PEPCK plays in gluconeogenesis may be mediated by the citric acid cycle, the activity of which was found to be directly related to PEPCK abundance.
PEPCK levels alone were not found to be highly correlated with gluconeogenesis in the mouse liver, as previous studies have suggested.Therefore, the role of PEPCK in gluconeogenesis may be more complex and involve more factors than was previously believed.
This gene belongs to the GPI family whose members encode multifunctional phosphoglucose isomerase proteins involved in energy pathways. The protein encoded by this gene is a dimeric enzyme that catalyzes the reversible isomerization of glucose-6-phosphate and fructose-6-phosphate.
glucose 6-phosphate <=> fructose 6-phosphate
The protein has different functions inside and outside the cell. In the cytoplasm, the protein is involved in glycolysis and gluconeogenesis, while outside the cell it functions as a neurotrophic factor for spinal and sensory neurons. The same protein is also secreted by cancer cells, where it is called autocrine motility factor and stimulates metastasis.Defects in this gene are the cause of nonspherocytic hemolytic anemia and a severe enzyme deficiency can be associated with hydrops fetalis, immediate neonatal death and neurological impairment.
What is cori cycle?
Cori cycle
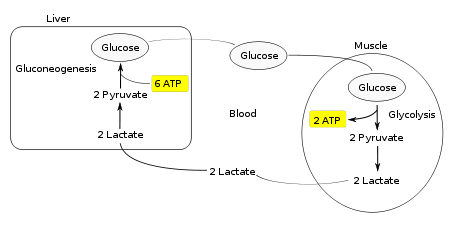
The Cori cycle, named after its discoverers, Carl Cori and Gerty Cori, refers to the metabolic pathway in which lactate produced by anaerobic glycolysis in the muscles moves to the liver and is converted to glucose, which then returns to the muscles and is converted back to lactate.Muscular activity requires energy, which is provided by the breakdown of glycogen in the skeletal muscles. The breakdown of glycogen, a process known as glycogenolysis, releases glucose in the form of glucose-6-phosphate (G-6-P). G-6-P is readily fed into glycolysis, a process that provides ATP to the muscle cells as an energy source. During muscular activity, the store of ATP needs to be constantly replenished. When the supply of oxygen is sufficient, this energy comes from feeding pyruvate, one product of glycolysis, into the Krebs cycle.
When oxygen supply is insufficient, typically during intense muscular activity, energy must be released through anaerobic respiration. Anaerobic respiration converts pyruvate to lactate by lactate dehydrogenase. Most important, fermentation regenerates NAD+, maintaining the NAD+ concentration so that additional glycolysis reactions can occur. The fermentation step oxidizes the NADH produced by glycolysis back to NAD+, transferring two electrons from NADH to reduce pyruvate into lactate. Refer to the main articles on glycolysis and fermentation for the details.
Instead of accumulating inside the muscle cells, lactate produced by anaerobic fermentation is taken up by the liver. This initiates the other half of the Cori cycle. In the liver, gluconeogenesis occurs. From an intuitive perspective, gluconeogenesis reverses both glycolysis and fermentation by converting lactate first into pyruvate, and finally back to glucose. The glucose is then supplied to the muscles through the bloodstream; it is ready to be fed into further glycolysis reactions. If muscle activity has stopped, the glucose is used to replenish the supplies of glycogen through glycogenesis.
Overall, the glycolysis part of the cycle produces 2 ATP molecules at a cost of 6 ATP molecules consumed in the gluconeogenesis part. Each iteration of the cycle must be maintained by a net consumption of 4 ATP molecules. As a result, the cycle cannot be sustained indefinitely. The intensive consumption of ATP molecules indicates that the Cori cycle shifts the metabolic burden from the muscles to the liver.The cycle's importance is based on the prevention of lactic acidosis in the muscle under anaerobic conditions. However, normally before this happens the lactic acid is moved out of the muscles and into the liver.
The cycle is also important in producing ATP, an energy source, during muscle activity. The Cori cycle functions more efficiently when muscle activity has ceased. This allows the oxygen debt to be repaid such that the Krebs cycle and electron transport chain can produce energy at peak efficiency.
Glycogenesis is the process of glycogen synthesis, in which glucose molecules are added to chains of glycogen for storage. This process is activated during rest periods following the Cori cycle, in the liver, and also activated by insulin in response to high glucose levels, for example after a carbohydrate-containing meal.
Glucose is converted into glucose-6-phosphate by the action of glucokinase or hexokinase.
Glucose-6-phosphate is converted into glucose-1-phosphate by the action of Phosphoglucomutase, passing through an obligatory intermediate step of glucose-1,6-bisphosphate.
Phosphoglucomutase (EC 5.4.2.2) is an enzyme that transfers a phosphate group on a glucose monomer from the 1' to the 6' position in the forward direction or the 6' to the 1' position in the reverse direction.
To be specific, it facilitates the interconversion of glucose 1-phosphate and glucose 6-phosphate.Phosphoglucomutase also acts in the opposite fashion when a large concentration of glucose-6-phosphate is present. In this case, it is the 1-carbon that is phosphorylated and the 6-carbon that is dephosphorylated. The resulting glucose-1-phosphate is then changed into UDP-glucose in a number of intermediate steps. If activated by insulin, glycogen synthase will proceed to clip the glucose from the UDP-glucose complex and on to the glycogen molecule.
Glucose-1-phosphate is converted into UDP-glucose by the action of Uridyl Transferase (also called UDP-glucose pyrophosphorylase) and pyrophosphate is formed, which is hydrolyzed by pyrophosphatase into 2 molecules of Pi.
UTP—glucose-1-phosphate uridylyltransferase also known as glucose-1-phosphate uridylyltransferase (or UDP–glucose pyrophosphorylase) is an enzyme associated with glycogenesis. It synthesizes UDP-glucose from glucose-1-phosphate and UTP; i.e.,
Glucose molecules are assembled in a chain by glycogen synthase, which must act on a pre-existing glycogen primer or glycogenin (small protein that forms the primer). The mechanism for joining glucose units is that glycogen synthase binds to UDPG, causing it to break down into an oxonium ion, also formed in glycogenolysis. This oxonium ion can readily add to the 4-hydroxyl group of a glucosyl residue on the 4 end of the glycogen chain.
Branches are made by branching enzyme (also known as amylo-α(1:4)->α(1:6)transglycosylase), which transfers the end of the chain onto an earlier part via α-1:6 glucosidic bond, forming branches, which further grow by addition of more α-1:4 glucosidic units.
Glycogenesis responds to hormonal control.
One of the main forms of control is the varied phosphorylation of glycogen synthase and glycogen phosphorylase. This is regulated by enzymes under the control of hormonal activity, which is in turn regulated by many factors. As such, there are many different possible effectors when compared to allosteric systems of regulation.
Epinephrine (Adrenaline)
Glycogen phosphorylase is activated by phosphorylation, whereas glycogen synthase is inhibited.
Glycogen phosphorylase is converted from its less active b form to an active a form by the enzyme phosphorylase kinase. This latter enzyme is itself activated by protein kinase A and deactivated by phosphoprotein phosphatase-1.
Protein kinase A itself is activated by the hormone adrenaline. Epinephrine binds to a receptor protein that activates adenylate cyclase. The latter enzyme causes the formation of cyclic AMP from ATP; two molecules of cyclic AMP bind to the regulatory subunit of protein kinase A, which activates it allowing the catalytic subunit of protein kinase A to dissociate from the assembly and to phosphorylate other proteins.
Returning to glycogen phosphorylase, the less active form (b) can itself be activated without the conformational change. 5'AMP acts as an allosteric activator, whereas ATP is an inhibitor, as already seen with phosphofructokinase control, helping to change the rate of flux in response to energy demand.
Epinephrine not only activates glycogen phosphorylase but also inhibits glycogen synthase. This amplifies the effect of activating glycogen phosphorylase. This inhibition is achieved by a similar mechanism, as protein kinase A acts to phosphorylate the enzyme, which lowers activity. This is known as co-ordinate reciprocal control. Refer to glycolysis for further information of the regulation of glycogenesis.
Insulin
Insulin has an antagonistic effect to adrenaline. When insulin binds on the G protein-coupled receptor, the alpha subunit of GDP in the G protein changes to GTP and dissociates from the inhibitory beta and gamma subunits. The alpha subunit binds on adenylyl cyclase to inhibit its activity. As a result, less cAMP then less protein kinase A will be produced. Thus, glycogen synthase, one of the targets of protein kinase A, will be in non-phosphorylated form, which is the active form of glycogen synthase. Active glycogen synthase can decrease the blood glucose level after a full meal
Calcium ions
Calcium ions or cyclic AMP (cAMP) act as secondary messengers. This is an example of negative control. The calcium ions activate phosphorylase kinase. This activates glycogen phosphorylase and inhibits glycogen synthase.
A glycogen branching enzyme is an enzyme that takes part in converting glucose to glycogen. It adds branches to the growing glycogen molecule. Glycogen is a branching polymer of large numbers of glucose units linked together. The structure is based on chains of glucose units with linkages between carbon atoms 1 and 4 of each pair of units (alpha 1, 4 linkages). These linkages are catalyzed by the enzyme glycogen synthase.Every 10 to 14 glucose units a side branch with an additional chain of glucose units occurs. The side chain attaches at carbon atom 6 of a glucose unit, and the linkage is termed an alpha-1,6 glycosidic bond. To form this connection a separate enzyme known as a branching enzyme is used. A branching enzyme attaches a string of seven glucose units to the sixth carbon of a glucose unit, usually in an interior location of the glycogen molecule.
This enzyme belongs to the family of transferases, to be specific, those glycosyltransferases that transfer hexoses (hexosyltransferases). The systematic name of this enzyme class is 1,4-alpha-D-glucan:1,4-alpha-D-glucan 6-alpha-D-(1,4-alpha-D-glucano)-transferase. Other names in common use include branching enzyme, amylo-(1,4→1,6)-transglycosylase, Q-enzyme, alpha-glucan-branching glycosyltransferase, amylose isomerase, enzymatic branching factor, branching glycosyltransferase, enzyme Q, glucosan transglycosylase, 1,4-alpha-glucan branching enzyme, plant branching enzyme, alpha-1,4-glucan:alpha-1,4-glucan-6-glycosyltransferase, and starch branching enzyme. This enzyme participates in starch and sucrose metabolism.
Glucose is an essential substrate for the metabolism of most cells. Because glucose is a polar molecule, transport through biological membranes requires specific transport proteins.
Active Transport - cotransporters
Transport of glucose through the apical membrane of intestinal and kidney epithelial cells depends on the presence of secondary active Na+/glucose symporters, SGLT-1 and SGLT-2, which concentrate glucose inside the cells, using the energy provided by cotransport of Na+ ions down their electrochemical gradient.
Passive transport - GLUTs
Facilitated diffusion of glucose through the cellular membrane is otherwise catalyzed by glucose carriers (protein symbol GLUT, gene symbol SLC2 for Solute Carrier Family 2) that belong to a superfamily of transport facilitators (major facilitator superfamily) including organic anion and cation transporters, yeast hexose transporter, plant hexose/proton symporters, and bacterial sugar/proton symporters. Molecule movement by such transporter proteins occurs by facilitated diffusion. This makes them energy-independent, unlike active transporters, which often require the presence of ATP to drive their translocation mechanism, and stall if the ATP/ADP ratio drops too low.
GLUTs are integral membrane proteins that contain 12 membrane-spanning helices with both the amino and carboxyl termini exposed on the cytoplasmic side of the plasma membrane. GLUT proteins transport glucose and related hexoses according to a model of alternate conformation,[1][2][3] which predicts that the transporter exposes a single substrate binding site toward either the outside or the inside of the cell. Binding of glucose to one site provokes a conformational change associated with transport, and releases glucose to the other side of the membrane. The inner and outer glucose-binding sites are, it seems, located in transmembrane segments 9, 10, 11;[4] also, the QLS motif located in the seventh transmembrane segment could be involved in the selection and affinity of transported substrate.[5][6]
Glucose transporter 1 (or GLUT1), also known as solute carrier family 2, facilitated glucose transporter member 1 (SLC2A1) is a protein that in humans is encoded by the SLC2A1 gene. GLUT1 facilitates the transport of glucose across the plasma membranes of mammalian cells.GLUT1 behaves as a Michaelis-Menten enzyme and contains 12 membrane-spanning alpha helices, each containing 20 amino acid residues. A helical wheel analysis shows that the membrane spanning alpha helices are amphipathic, with one side being polar and the other side hydrophobic. Six of these membrane spanning helices are believed to bind together in the membrane to create a polar channel in the center through which glucose can traverse, with the hydrophobic regions on the outside of the channel adjacent to the fatty acid tails of the membrane.
Energy-yielding metabolism in erythrocytes depends on a constant supply of glucose from the blood plasma, where the glucose concentration is maintained at about 5mM. Glucose enters the erythrocyte by facilitated diffusion via a specific glucose transporter, at a rate about 50,000 times greater than uncatalyzed transmembrane diffusion. The glucose transporter of erythrocytes (called GLUT1 to distinguish it from related glucose transporters in other tissues) is a type III integral protein with 12 hydrophobic segments, each of which is believed to form a membrane-spanning helix. The detailed structure of GLUT1 is not known yet, but one plausible model suggests that the side-by-side assembly of several helices produces a transmembrane channel lined with hydrophilic residues that can hydrogen-bond with glucose as it moves through the channel.
GLUT1 is responsible for the low-level of basal glucose uptake required to sustain respiration in all cells. Expression levels of GLUT1 in cell membranes are increased by reduced glucose levels and decreased by increased glucose levels.
GLUT1 is also a major receptor for take-up of Vitamin C as well as glucose, especially in non vitamin C producing mammals as part of an adaptation to compensate by participating in a Vitamin C recycling process. In mammals that do produce Vitamin C, GLUT4 is often expressed instead of GLUT1.
Glucose transporter 2 (GLUT2) also known as solute carrier family 2 (facilitated glucose transporter), member 2 (SLC2A2) is a transmembrane carrier protein that enables passive glucose movement across cell membranes. It is the principal transporter for transfer of glucose between liver and blood, and for renal glucose reabsorption. In humans, this protein is encoded by the SLC2A2 gene.
GLUT2 is found in cellular membranes of:
liver
pancreatic beta cells
hypothalamus
basolateral and brush border membrane of small intestine
basolateral membrane of renal tubular cells
Defects in the SLC2A2 gene are associated with a particular type of glycogen storage disease called Fanconi-Bickel syndrome.
It has been proposed by genetics researchers in neonatal and maternal-fetal medicine at Harvard University Medical School and Beth-Israel Deaconess Hospital Medical Center that this creates a problem for drug-treated diabetic pregnancies in which glucose levels in the woman are uncontrolled, exposing her fetus to the possibility of neural tube and cardiac defects in the early-developing brain, spine, and heart. However, whilst a lack of GLUT2 adaptability is negative, it is important to remember the fact that the main result of untreated gestational diabetes appears to cause babies to be of above-average size, which may well be an advantage that is managed very well with a healthy GLUT2 status.
Maintaining a regulated osmotic balance of sugar concentration between the blood circulation and the interstitial spaces is critical in some cases of edema including cerebral edema.
GLUT2 appears to be particularly important to osmoregulation, and preventing edema-induced stroke, transient ischemic attack or coma, especially when blood glucose concentration is above average. GLUT2 could reasonably referred to as the "diabetic glucose transporter" or a "stress hyperglycemia glucose transporter."
GLUT3 is a high-affinity isoform of Type I glucose transporter that is mostly expressed in neurons, where it is believed to be the main glucose transporter isoform. It is also expressed in the placenta.
Glucose transporter type 4, also known as GLUT4, is a protein that in humans is encoded by the GLUT4 gene. GLUT4 is the insulin-regulated glucose transporter found in adipose tissues and striated muscle (skeletal and cardiac) that is responsible for insulin-regulated glucose translocation into the cell. This protein is expressed only in muscle and fat cells, the major tissues in the body that respond to insulin. The first evidence for this distinct glucose transport protein was provided by David James in 1988.
GLUT4 is primarily found in:
Skeletal muscle
Cardiac muscle
Adipose tissue
In the absence of insulin, GLUT4 is sequestered in the interior of muscle and fat cells within lipid bilayers of vesicles. Insulin induces the translocation of GLUT4 from intracellular storage sites to the plasma membrane. Insulin binds to the insulin receptor in its dimeric form. The receptor phosphorylates and subsequently activates IRS-1, which in turn binds the enzyme PI-3 kinase which converts the membrane lipid PIP2 to PIP3. PIP3 generates a binding site for PKB (protein kinase B), and also for PDK1 which, being localized together with PKB, can phosphorylate and activate PKB. Once phosphorylated, PKB is in its active form and phosphorylates TBC1D4, which inhibits the GTPase-activating domain associated with TBC1D4. Inhibition of the GTPase-activating domain leaves proteins next in the cascade in their active form and stimulates GLUT4 to be expressed on the plasma membrane.
At the cell surface, GLUT4 permits the facilitated diffusion of circulating glucose down its concentration gradient into muscle and fat cells. Once within cells, glucose is rapidly phosphorylated by glucokinase in the liver and hexokinase in other tissues to form glucose-6-phosphate, which then enters glycolysis or is polymerized into glycogen. Glucose-6-phosphate cannot diffuse back out of cells, which also serves to maintain the concentration gradient for glucose to passively enter cells.
Glycogen phosphorylase was the first allosteric enzyme to be discovered.[7] This accomplishment was one of many landmark achievements made by Carl and Gerty Cori. In 1943, with the help of Arda Green, the pair illustrated that glycogen phosphorylase existed in either the a or b forms depending on its phosphorylation state, as well as in the R or T states based on the presences of AMP.[8]
The glycogen phosphorylase monomer is a large protein, composed of 842 amino acids with a mass of 97.434 kDa in muscle cells. While the enzyme can exist as an inactive monomer or tetramer, it is biologically active as a dimer of two identical subunits.[9]
R and T States of Glycogen Phosphorylase b Tower Helices, on the left and right respectively. Note the relative positioning of the central tower helices, as well as the increased interactions between subunits in the R state. PDB3CEH, PDB3E3O
The glycogen phosphorylase dimer has many regions of biological significance, including catalytic sites, glycogen binding sites, allosteric sites, and a reversibly phosphorylated serine residue. First, the catalytic sites are relatively buried, 15Å from the surface of the protein and from the subunit interface.[10] This lack of easy access of the catalytic site to the surface is significant in that it makes the protein activity highly susceptible to regulation, as small allosteric effects could greatly increase the relative access of glycogen to the site.
Perhaps the most important regulatory site is Ser14, the site of reversible phosphorylation very close to the subunit interface. The structural change associated with phosphorylation, and with the conversion of phosphorylase b to phosphorylase a, is the arrangement of the originally disordered residues 10 to 22 into α helices. This change increases phosphorylase activity up to 25% even in the absence of AMP, and enhances AMP activation further.[11]
The allosteric site of AMP binding on muscle isoforms of glycogen phosphorylase are close to the subunit interface just like Ser14. Binding of AMP at this site, corresponding in a change from the T state of the enzyme to the R state, results in small changes in tertiary structure at the subunit interface leading to large changes in quaternary structure.[7] AMP binding rotates the tower helices (residues 262-278) of the two subunits 50˚ relative to one another through greater organization and intersubunit interactions. This rotation of the tower helices leads to a rotation of the two subunits by 10˚ relative to one another, and more importantly disorders residues 282-286 (the 280s loop) that block access to the catalytic site in the T state but do not in the R state.[10]
The final, perhaps most curious site on the glycogen phosphorylase protein is the so-called glycogen storage site. Residues 397-437 form this structure, which allows the protein to covalently bind to the glycogen chain a full 30 Å from the catalytic site . This site is most likely the site at which the enzyme binds to glycogen granules before initiating cleavage of terminal glucose molecules. In fact, 70% of dimeric phosphorylase in the cell exists as bound to glycogen granules rather than free floating.[12]
In mammals, the major isozymes of glycogen phosphorylase are found in muscle, liver, and brain. The brain type is predominant in adult brain and embryonic tissues, whereas the liver and muscle types are predominant in adult liver and skeletal muscle, respectively.[13]
The overall reaction of Glycogen phosphorylase is written as:
Although the reaction is reversible in solution, within the cell the enzyme only works in the forward direction as shown above because the concentration of inorganic phosphate is much higher than that of glucose-1-phosphate.[14]
Action of Glycogen Phosphorylase on Glycogen
Glycogen phosphorylase can act only on linear chains of glycogen (α1-4 glycosidic linkage). Its work will immediately come to a halt four residues away from α1-6 branch (which are exceedingly common in glycogen). In these situations, a debranching enzyme is necessary, which will straighten out the chain in that area. In addition, the enzyme transferase shifts a block of 3 glucosyl residues from the outer branch to the other end, and then a α1-6 glucosidase enzyme is required to break the remaining (single glucose) α1-6 residue that remains in the new linear chain. After all this is done, glycogen phosphorylase can continue. The enzyme is specific to α1-4 chains, as the molecule contains a 30-angstrom-long crevice with the same radius as the helix formed by the glycogen chain; this accommodates 4-5 glucosyl residues, but is too narrow for branches. This crevice connects the glycogen storage site to the active, catalytic site.
Glycogen phosphorylase has a pyridoxal phosphate (PLP, derived from Vitamin B6) at each catalytic site. Pyridoxal phosphate links with basic residues (in this case Lys680) and covalently forms a Schiff base. Once the Schiff base linkage is formed, holding the PLP molecule in the active site, the phosphate group on the PLP readily donates a proton to an inorganic phosphate molecule, allowing the inorganic phosphate to in turn be deprotonated by the oxygen forming the α-1,4 glycosidic linkage. PLP is readily deprotonated because its negative charge is not only stabilized within the phosphate group, but also in the pyridine ring, thus the conjugate base resulting from the deprotonation of PLP is quite stable. The protonated oxygen now represents a good leaving group, and the glycogen chain is separated from the terminal glycogen in an SN1 fashion, resulting in the formation of a glucose molecule with a secondary carbocation at the 1 position. Finally, the deprotonated inorganic phosphate acts as a nucleophile and bonds with the carbocation, resulting in the formation of glucose-1-phosphate and a glycogen chain shortened by one glucose molecule.
There is also an alternative proposed mechanism involving a positively charged oxygen in a half-chair conformation. [15]
The inhibition of glycogen phosphorylase has been proposed as one method for treating type 2 diabetes.[16] Since glucose production in the liver has been shown to increase in type 2 diabetes patients,[17] inhibiting the release of glucose from the liver’s glycogen’s supplies appears to be a valid approach. The cloning of the human liver glycogen phosphorylase (HLGP) revealed a new allosteric binding site near the subunit interface that is not present in the rabbit muscle glycogen phosphorylase (RMGP) normally used in studies. This site was not sensitive to the same inhibitors as those at the AMP allosteric site,[18] and most success has been had synthesizing new inhibitors that mimic the structure of glucose, since glucose-6-phosphate is a known inhibitor of HLPG and stabilizes the less active T-state.[19] These glucose derivatives have had some success in inhibiting HLPG, with predicted Ki values as low as 0.016 mM.[20]
Mutations in the muscle isoform of glycogen phosphorylase (PYGM) are associated with McArdle's Disease (glycogen storage disease type V). More than 65 mutations in the PYGM gene that lead to McArdle disease have been identified to date.[21][22] Symptoms of McArdle disease include muscle weakness, myalgia, and lack of endurance, all stemming from low glucose levels in muscle tissue.[23]
Mutations in the liver isoform of glycogen phosphorylase (PYGL) are associated with Hers' Disease (glycogen storage disease type VI).[24][25] Hers' disease is often associated with mild symptoms normally limited to hypoglycemia, and is sometimes difficult to diagnose due to residual enzyme activity.[26]
The brain isoform of glycogen phosphorylase (PYGLB) has been proposed as a biomarker for gastric cancer.[27]
↑Oka Y, Asano T, Shibasaki Y, Lin J, Tsukuda K, Katagiri H, Akanuma Y, Takaku F (1990). "C-terminal truncated glucose transporter is locked into an inward-facing form without transport activity". Nature. 345 (6275): 550–3. doi:10.1038/345550a0. PMID2348864.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑Hebert D, Carruthers A (1992). "Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1". J. Biol. Chem. 267 (33): 23829–38. PMID1429721.
↑Cloherty E, Sultzman L, Zottola R, Carruthers A (1995). "Net sugar transport is a multistep process. Evidence for cytosolic sugar binding sites in erythrocytes". Biochemistry. 34 (47): 15395–406. doi:10.1021/bi00047a002. PMID7492539.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑Hruz P, Mueckler M (2001). "Structural analysis of the GLUT1 facilitative glucose transporter (review)". Mol. Membr. Biol. 18 (3): 183–93. doi:10.1080/09687680110072140. PMID11681785.
↑Seatter M, De la Rue S, Porter L, Gould G (1998). "QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site". Biochemistry. 37 (5): 1322–6. doi:10.1021/bi972322u. PMID9477959.{{cite journal}}: CS1 maint: multiple names: authors list (link)
↑Hruz P, Mueckler M (1999). "Cysteine-scanning mutagenesis of transmembrane segment 7 of the GLUT1 glucose transporter". J. Biol. Chem. 274 (51): 36176–80. doi:10.1074/jbc.274.51.36176. PMID10593902.
↑ abJohnson LN, Barford, D (1990). "Glycogen phosphorylase. The structural basis of the allosteric response and comparison with other allosteric proteins". Journal of Biological Chemistry. 265 (5): 2409–2412. PMID2137445. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑ abJohnson LN (1992). "Glycogen phosphorylase: control by phosphorylation and allosteric effectors". FASEB Journal. 6 (6): 2274–82. PMID1544539. {{cite journal}}: Unknown parameter |month= ignored (help)
↑Newgard CB, Hwang PK, Fletterick, RJ (1989). "The family of glycogen phosphorylases: structure and function". Critical Reviews Biochemistry and Molecular Biology. 24 (1): 69–99. doi:10.3109/10409238909082552. PMID2667896. {{cite journal}}: Cite has empty unknown parameter: |month= (help)CS1 maint: multiple names: authors list (link)
↑Meyer F, Heilmeyer LM Jr, Haschke RH, Fischer EH (1970). "Control of phosphorylase activity in a muscle glycogen particle. I. Isolation and characterization of the protein-glycogen complex". Journal of Biological Chemistry. 245 (24): 6642–6648. PMID4320610. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑David ES, Crerar MM (1986). "Quantitation of muscle glycogen phosphorylase mRNA and enzyme amounts in adult rat tissues". Biochim. Biophys. Acta. 880 (1): 78–90. PMID3510670. {{cite journal}}: Unknown parameter |month= ignored (help)
↑ abLivanova NB, Chebotareva NA, Eronina TB, Kurganov BI (2002). "Pyridoxal 5′_Phosphate as a Catalytic and Conformational Cofactor of Muscle Glycogen Phosphorylase b". Biochemistry (Moscow). 67 (10): 1089–1998. doi:10.1023/A:1020978825802. PMID12460107. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Palm D, Klein HW, Schinzel R, Buehner M, Helmreich, EJM (1990). "The role of pyridoxal 5'-phosphate in glycogen phosphorylase catalysis". Biochemistry. 29 (5): 1099–1107. doi:10.1021/bi00457a001. PMID2182117. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Somsák L, Nagya V, Hadady Z, Docsa T, Gergely P. (2003). "Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: recent developments". Current Pharmacological Design. 9 (15): 1177–89. doi:10.2174/1381612033454919. PMID12769745. {{cite journal}}: Cite has empty unknown parameter: |month= (help)CS1 maint: multiple names: authors list (link)
↑Moller DE (2001). "New drug targets for type 2 diabetes and the metabolic syndrome". Nature. 414 (6865): 821–7. doi:10.1038/414821a. PMID11742415. {{cite journal}}: Unknown parameter |month= ignored (help)
↑Oikonomakos NG, Kontou M, Zographos SE, Tsitoura HS, Johnson LN, Watson KA, Mitchell EP, Fleet GW, Son JC, Bichard CJ; et al. (1994). "The design of potential antidiabetic drugs: experimental investigation of a number of beta-D-glucose analogue inhibitors of glycogen phosphorylase". European Journal of Drug Metabolism and Pharmacology. 19 (3): 185–92. doi:10.1007/BF03188920. PMID7867660. {{cite journal}}: Explicit use of et al. in: |author= (help); Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Hopfinger A J, Reaka A, Venkatarangan P, Duca J S, Wang S. (1999). "Prediction of Ligand−Receptor Binding Free Energy by 4D-QSAR Analysis: Application to a Set of Glucose Analogue Inhibitors of Glycogen Phosphorylase". Journal of Chemical Information and Computer Science. 39: 1141–1150. doi:10.1021. {{cite journal}}: Check |doi= value (help); Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Chang S, Rosenberg MJ, Morton H, Francomano CA, Biesecker LG (1998). "Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI". Hum. Mol. Genet. 7 (5): 865–70. doi:10.1093/hmg/7.5.865. PMID9536091. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Tang NL, Hui J, Young E, Worthington V, To KF, Cheung KL, Li CK, Fok TF (2003). "A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity". Molecular Genetics and Metabolism. 79 (2): 142–145. doi:10.1016/S1096-7192(03)00068-4. PMID12809646. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)
↑Shimada S, Matsuzaki H, Marutsuka T, Shiomori K, Ogawa M (2001). "Gastric and intestinal phenotypes of gastric carcinoma with reference to expression of brain (fetal)-type glycogen phosphorylase". J. Gastroenterol. 36 (7): 457–64. doi:10.1007/s005350170068. PMID11480789. {{cite journal}}: Unknown parameter |month= ignored (help)CS1 maint: multiple names: authors list (link)